
 Star fruit neurotoxin identified
Star fruit neurotoxin identified
Neurotoxin from Star Fruit
Patients with kidney disease have to watch what they eat: bananas, oranges, tomatoes, nuts, broccoli, and beans are all off-limits. Putting star fruit or carambola on the menu would be downright dangerous. This fruit contains a substance that is a deadly neurotoxin for people with kidney disease. Brazilian researchers have now isolated and identified this neurotoxin. As they report in the journal Angewandte Chemie, it is an amino acid similar to phenylalanine.
Caramboxin: Patients suffering from chronic kidney disease are frequently intoxicated after ingesting star fruit. The main symptoms of this intoxication are named in the picture. Bioguided chemical procedures resulted in the discovery of caramboxin, which is a new phenylalanine-like molecule that is responsible for intoxication. Functional experiments in vivo and in vitro point towards the glutamatergic ionotropic molecular actions of caramboxin, which explains its convulsant and neurodegenerative properties.
Sweet Poison
http://www.chemistryviews.org/details/ezine/5542471/Sweet_Poison.html
star fruit (Averrhoa carambola) or carambola has been cultivated in Malaysia, Southern China, Taiwan, India and Brazil. It is rather popular in the Philippines and Queensland, Australia and moderately so in some of the South Pacific Islands, particularly Tahiti, New Caledonia and Netherlands New Guinea, Guam and in Hawaii and south Florida. There are some subspecies in the Caribbean Islands, in Central America and in tropical West Africa. The fruits are also available in many European countries and Canada. Range of soluble oxalate salts concentrations obtained from many cultivars varies from 80 to 730 mg/100 g of the fruit
Structural Elucidation and Spectroscopic Data of Caramboxin (1)
Most 1H and 13C NMR data from the isolated compound were easily assigned due to the relationship
of this compound with well-known aromatic amino acids such as phenylalanine. The side chain is
identical to phenylalanine as attributed in the NMR data below. The tetra-substituted pattern of the aromatic ring was also easily recognized by the only two signals for aromatic protons at δ 6.42 and6.37 with a coupling constant (2.0 Hz) typical of aromatic meta coupling. The positioning of
substituents in the aromatic ring were attributed due to the 13C chemical shifts and confirmed by
HMBC experiment. In this way acetyl group was placed at C-6 due to the low chemical shift (104.1ppm) of this aromatic carbon; methoxyl was placed at C-3 due to its highest chemical shift (165.2ppm) among aromatic protons, which was confirmed by HMBC experiment; finally the hydroxyl group was placed at the remaining C-5. Comparison of the observed accurate mass measurement with theoretically calculated formulae for the signal at m/z 256.0823 allows only a few reasonable [MH]+ ion formulae within a standard deviation of 50 ppm. The ion formula [C11H13NO6 + H]+ had the best mass accuracy (0.7 ppm error) and correlation with the NMR spectra. The MS/MS spectrum of m/z 256 shows an intense ion at m/z 192 in addition to neutral elimination of H2O (m/z 238) and CO2 (m/z 212) from the carboxylic acid group. In source dissociation followed by MS/MS analysis revealed that m/z 192 was only obtained from m/z 238, due to the elimination of CH2O2 (46 massunits) as a neutral molecule by 1,2 elimination. The same mechanism was also observed for the m/z166 formation from the ion at m/z 212. Finally, 2D NMR data from HMQC and HMBC confirmed the entire spectral assignment
Caramboxin: 1H-NMR (400 MHz, D2O) δ 6.42 (d, J = 2.0 Hz, 1H, H-4), 6.37 (d, J = 2.0 Hz, 1H, H-2), 4.25 (dd, J = 5.5, 8.0 Hz, 1H, H-8), 3.80 (s, 3H, H-11), 3.66 (dd, J = 14.0, 5.5 Hz, 1H, H-7A),3.18 (dd, J = 14.0, 8.0 Hz, 1H, H-7B);
13C-NMR (100 MHz, DMSO-d6) δ 172.8 (C, C-9), 171.2 (C,C-10), 165.2 (C, C-5), 162.2 (C, C-3), 138.8 (C, C-1), 110.0 (CH, C-2), 104.1 (C, C-6), 100.5 (CH,C-4), 55.4 (CH3, C-11), 53.5 (CH, C-8), 35.9 (CH2, C-7); 15N-NMR (50 MHz, DMSO-d6) δ -268(nitromethane as internal reference).
HMBC (500 MHz, DMSO-d6): H-2 → C-3, C-4, C-7; H-4 →C-2, C-3, C-5; H-7 → C-1, C-2, C-8, C-9; H-8 → C-1, C-7, C-9; H-11 → C-3;
HRMS (m/z): [MH]+calcd for C11H14NO6, 256.0816; found: 256.0818






Whisky LactoneWhisky lactone, also known as β-methyl-γ-octalactone or quercus lactone (from the Latin for oak treeQuercus alba), is a flavouring found in American bourbon whiskies, and is also found in all types of oak. The flavour gets into the whisky when it’s matured in oak barrels. The pure molecule has a fierce, strong, and sweet smell and can be dissolved in alcohol in any proportion.
6 http://www.sciencedirect.com/science/article/pii/S0957416698000809 5-butyl-4-methyloxolan-2-one | CAS Registry Number: 39212-23-2
The cis isomer is the chemical extracted from oak wood that gives whiskey a coconut-like aroma. But not all isomers of this molecule are quite this tasty. The trans isomers of 3-methyl-4-octanolide is by contrast, used as an insect repellent.
Physical data of the obtained cis-whiskey lactone are
|






http://www.chemistryviews.org/details/ezine/5392681/Amide_Hydrogenation_in_Flow_Reactor.html
Amide Hydrogenation in Flow Reactor (wiley)
Amines are produced by amide hydrogenation over a bimetallic platinum–rhenium catalyst in a high-throughput vertical flow reactor
 A library of novel 5-nitroimidazole antibiotics displayed broad-spectrum activity
A library of novel 5-nitroimidazole antibiotics displayed broad-spectrum activity
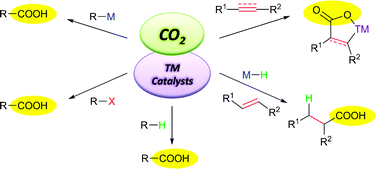

Carbon dioxide is an important carbon source in the atmosphere and is “problematic” toward the activities of human beings. Although carbon dioxide is a cheap, abundant and relatively nontoxic C1 source, its chemical transformations have not been widely developed so far and are still far from synthetic applications, especially in the construction of the C–C bond. This critical review summarizes the recent advances on transition-metal-catalyzed C–C bond formation through the fixation of carbon dioxide and their synthetic applications (124 references).
http://pubs.rsc.org/en/content/articlelanding/2011/cs/c0cs00129e#!divAbstract
Transition-metal-catalyzed C–C bond formation through the fixation ofcarbon dioxide
Corresponding authors
DOI: 10.1039/C0CS00129E





The N-methylation of electron-deficient pyrroles was affected using dimethyl carbonate in the presence of DMF and catalytic DABCO. This alkylation methodology has proven useful for the alkylation of a variety of pyrroles in 72−98% yields and is considered to be greenchemistry relative to the more common use of methyl halides or dimethyl sulfate.

The use of alternative reaction solvents is reviewed in terms of life cycle. Supercritical CO2, ionic liquids, fluorous solvents, water, and renewable organics are compared on the basis of their solvency, ease of use, reusability, health and safety, environmental impact, and economic cost.
James H. Clark * and Stewart J. Tavener
Green Chemistry Centre, Department of Chemistry, University of York, Heslington, York, U.K. YO10 5DD
Org. Process Res. Dev., 2007, 11 (1), pp 149–155
DOI: 10.1021/op060160g
Publication Date (Web): November 4, 2006
http://pubs.acs.org/doi/full/10.1021/op060160g?prevSearch=GREEN%2BSOLVENTS&searchHistoryKey=
This article critically reviewS the use of alternative solvents in chemistry. Rather than follow the well-trodden path of discussing in turn the reactions that have been performed in each major type of alternative solvent, we will instead structure our article in terms of what we consider to be the fundamental issues: life cycle analysis (so as to establish the “green” and sustainability aspects from the outset), solvency (so as to consider what is needed in the application and how the alternatives manage to meet these needs), and application (to consider practical issues in both process and product).

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
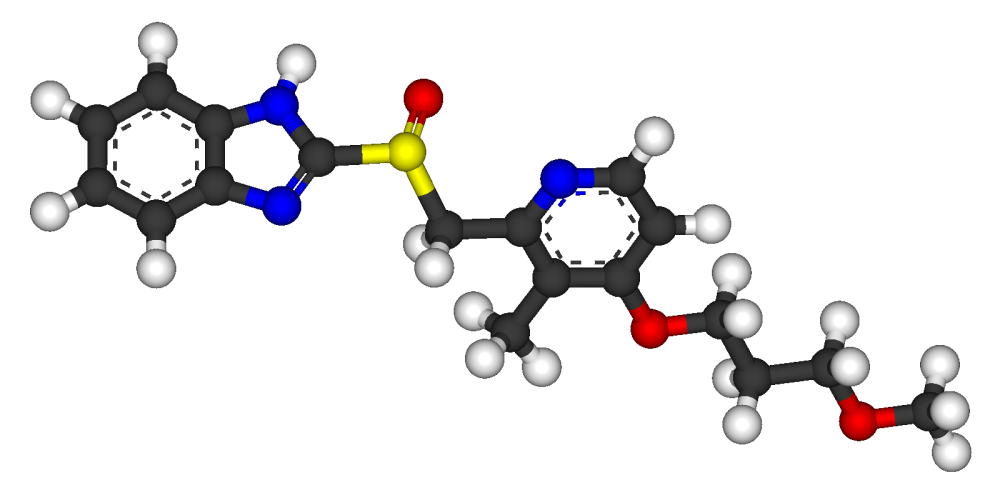
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
- US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.



Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
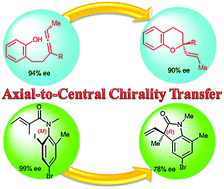
Axial-to-central chirality transfer in cyclization processes
Substrates, bearing axial chirality, can cyclize intra- or inter-molecularly with concomitant transfer of axial-to-central chirality to produce at least one stereocenter. In order to satisfy a strict definition of axial-to-central chirality transfer, the initial axial chirality must be lost during the cyclization process. Highly functionalized enantiopure carbocycles and heterocycles were prepared using this strategy. The transformations of configurationally stable substrates take place with high regio- and stereo-selectivity. Selected examples involving allenes, biaryls, arylamides and transient axially chiral short-lived species are discussed. Special attention is focused on the mechanistic rationale of the chirality transfer.
DOI: 10.1039/C3CS60182J