

Damiana contains a glycoside compound called arbutin. In the urinary tract, arbutin is converted into a chemical called hydroquinone. In large amounts, hydroquinone can cause nausea, vomiting, tinnitus (ringing in the ears, convulsions, and eventually, collapse and death.
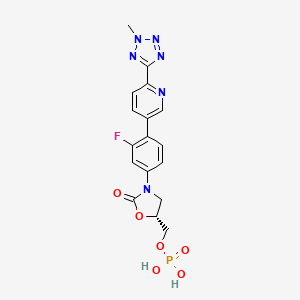
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
……………………………..
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
…………………………………………………….
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,

These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
…………………………………………………………..
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
…………………………………………..
imp patents
| US2010305069 | 12-3-2010 | OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE |
| US7816379 | 10-20-2010 | Oxazolidinone derivatives |
| US2009192197 | 7-31-2009 | NOVEL OXAZOLIDINONE DERIVATIVES |
…………………………………………..
TEDIZOLID disodium salt
59 nos in
http://www.google.com/patents/US20130102523

 38 nos
38 nos
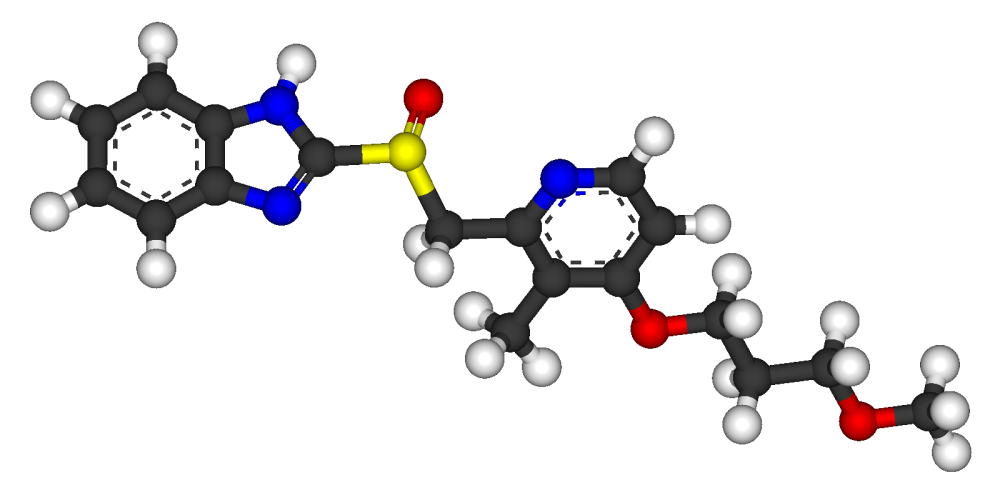
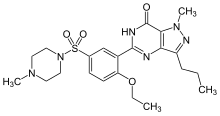
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

…………………………………………………………………………………………………………………………………………………………….
Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
……………………………………
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
………………………………………………………………………………………………………………………..
SYNTHESIS

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

………………………………………………………………………………………………………………………….
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
………………………………………………………………………………………………………………………………………………………….
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
…………………………………………………………………………………………………………………………………………..
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

………………………………………………………………………………………………………………………………………………………
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
………………………………………………………
IMPURITIES
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |  |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |  |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |  |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |  |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |
  |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |  |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |  |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |  |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |  |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |  |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |  |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
……………………………………………..
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
……………………………………………………………………………………….


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007
Share this:





]]>
 A library of novel 5-nitroimidazole antibiotics displayed broad-spectrum activity
A library of novel 5-nitroimidazole antibiotics displayed broad-spectrum activity
Share this:
]]>

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
Rabeprazole Sodium is commercially available in a pharmaceutical composition under the brand name ACIPHEX® marketed by Eisai and is covered under US 5045552 (JP priority application No. JP1987002198919870202; JP1987007778419870331; JP1986027053619861113
- US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.

The condensation of 4-chloro-2,3-dimethylpyridine N-oxide (I) with 3-methoxypropanol (II) by means of NaH in DMSO gives 4-(3-methoxypropoxy)-2,3-dimethylpyridine N-oxide (III), which is treated with acetic anhydride at 90 C yielding 2-(acetoxymethyl)-4-(3-methoxypropoxy)-3-methylpyridine (IV). The hydrolysis of (IV) with NaOH in ethanol affords 2-(hydroxymethyl)-4-(3-methoxypropoxy)-3-methylpyridine (V), which by treatment with SOCl2 in dichloromethane is converted into 2-(chloromethyl)-4-(3-methoxypropoxy)-3-methylpyridine (VI). The condensation of (VI) with 2-mercaptobenzimidazole (VII) by means of NaOH in ethanol gives 2-[4-(3-methoxypropoxy)-3-methylpyridin-2-ylmethylthio]benzimidazole (VIII), which is oxidized with m-chloroperbenzoic acid in ether – dichloromethane to afford 2-[4-(3-methoxypropoxy)-3-methylpyridin-2-ylsulfinyl]benzimidazole (IX). Finally, this compound is treated wtih aqueous Na2CO3.
NEXT………………….

A synthesis for [14C]-labeled E-3810 has been described: The cyclization of o-phenylenediamine (I) with [14C]-labeled carbon disulfide (II) by means of aqueous KOH gives the potassium salt of [14C]-2-mercaptobenzimidazole (III), which is treated with acetic acid to obtain the corresponding thiol (IV). The condensation of (IV) with 2-(chloromethyl)-4-(3-methoxypropoxy)-3-methylpyridine (V) by means of NaOH in refluxing ethanol yields [14C]-labeled 2-[4-(3-methoxypropoxy)-3-methylpyridin-3-ylmethylthio]benzimidazole (VI). The oxidation of (VI) with m-chloroperbenzoic acid in dichloromethane affords the corresponding sulfoxide (VIII), which is finally treated with 0.1 N NaOH in ethanol.

References1:
Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
References2:
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
References3:
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
Share this:
]]>

DR ANTHONY MELVIN CRASTO Ph.D
WORLDDRUGTRACKER
ANNOUNCING ONE LAKH PLUS VIEWS ON ALL BLOGS- DR ANTHONY CRASTO
- Eurekamoments in Organic Chemistry
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- Drug Scaleup and Manufacturing International
SEE ALSO
- Organic Chemistry by Dr Anthony
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India, in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now sSanofi, Searle India ltd, now Rpg lifesciences, etc. he is now helping millions, has million hits on google on all organic chemistry websites. His New Drug Approvals, Green Chemistry International, Eurekamoments in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide . He gas good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, polymorphism etc He suffered a paralytic stroke in dec 2007 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Personal Links
- my sites on the net
- DR ANTHONY MELVIN CRASTO
- GOOGLE GROUP ORGANIC PROCESS DEVELOPMENT
- mixxt
- epernicus
- scipeople
- jimdo
- yolasite
- my cv
- slidestaxx
- wordpress blog
- ABOUT ME
- BRANDSITE
- SKILLPAGES
- Academia.edu
- RESEARCHGATE
- DIIGO
- SLIDESHATE
- WIX
- WIX BLOG
- ISSUU
- SCRIBD
- BIZ
- GOOGLE BLOG
- APNACIRCLE
- Eurekamoments in Organic Chemistry
- Organic Chemistry by Dr Anthony
- Green Chemistry International
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
- DR ANTHONY MELVIN CRASTO Ph.D
- Pharmaceuticals
- Medicinal chemistry
- Organic chemistry literature
- Patent related site
- Green chemistry
- Reagents
- R & D
- Molecules
- Heterocyclic chem
- Sourcing
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- GOOGLE PLUS
- Drug Scaleup and Manufacturing International
amcrasto@gmail.com
email me if u like my posts
Share this:
]]>

Zeolites modified with aminoalkoxysilanes and cyclodextrin show different uptake and release properties depending on which silane is used
Multifunctional nanocontainers for imaging, targeting, and drug release are a main research area in bio-nanomedicine. Jurriaan Huskens and colleagues, University of Twente, The Netherlands, have functionalized nanoporous zeolite L crystals with β-cyclodextrin (CD) to give multifunctional systems that have the potential for encapsulation of drug molecules inside the zeolite pores and noncovalent attachment of other, for example, targeting, ligand molecules on its surface.
Share this:
]]>
Share this:
]]>
READ ALL AT
http://newdrugapprovals.wordpress.com/2013/09/14/benazepril-hydrochloride-synthesis-and-review/
BENAZEPRIL HYDROCHLORIDE SYNTHESIS
Share this:
]]>
click above for all you want to know about drugs
by
DR ANTHONY MELVIN CRASTO Ph.D , Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues,
Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India.
Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now Sanofi Aventis, & Searle India ltd, now Rpg lifesciences, etc. He has worked in Basic research, Neutraceuticals, Natural products, Flavors, Fragrances, Pheromones, Vet Drugs, Drugs, formulation, GMP etc. He has total 25 yrs exp in this field, he is now helping millions, has million hits on google on all organic chemistry websites.
His New Drug Approvals , Green Chemistry International, Eurekamoments in Organic Chemistry , Organic Chemistry by Dr Anthony, WIX BLOG , are some most read chemistry blogs
He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide .
He has good proficiency in Technology Transfer, Spectroscopy , Stereochemistry , Synthesis, Reactions in Org Chem , Polymorphism, Pharmaceuticals , Medicinal chemistry , Organic chemistry literature , Patent related site , Green chemistry , Reagents , R & D , Molecules , Heterocyclic chem , Sourcing etc
He suffered a paralytic stroke in dec 2006 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Share this:
]]>
CHECK OUT MY BLOG NEW DRUG APPROVALS AT
https://newdrugapprovals.wordpress.com/
sildanafil
Erection of the penis in males is often a result of a state of sexual arousal. Erectile dysfunction occurs when it becomes difficult to produce erection even in a state of adequate arousal. Erectile dysfunction can occur at any age to any one and at any point of time. It can be due to a vast array of reasons, ranging from fatigue to serious diabetic or heart conditions. While causes like fatigue can be taken care of by simple rest and a good night’s sleep, serious causes like diabetes and cardiovascular diseases can be a little difficult to deal with. Erectile dysfunction does not necessarily mean that there is something physically wrong within the body, as it can also be a result of a vast number of psychological reasons. The loss of erection in itself can give rise to a vast number of psychological problems like loss of self respect and confidence and, hence, requires immediate medical assistance.

vardenafil
You can characterize erectile dysfunction (also known as the problem of male impotency) into two broad categories: firstly, when sometimes full erections are obtained, like when the person under consideration is in deep sleep. This condition is due to the failure of getting an erection due to a psychological reason and can be solved with professional psychological assistance. Secondly, when no erection is obtained. This is generally when the physical structure is not working properly.

tadalafil
Erectile dysfunction takes place when a man fails to get a proper erection or is not able to sustain it to indulge in sexual intercourse. There is no formal means of detecting and diagnosing an erectile dysfunction. However, blood tests are conducted in such cases as they generally give a fair idea of the underlying diseases such as prolactinoma, diabetes and hypogonadism. Impotency is generally a result of poor health conditions and can be a result of obesity or malnutrition. There are a number of tests along with the blood tests that are undertaken to determine the nature and extent of an erectile dysfunction problem. These are duplex ultrasound to evaluate the blood flow, penile nervous function test such as bulbocavernosus reflex, nocturnal penile tumescence, penile biothesiometry, Magnetic Resonance Angiography (MRA), etc.
avanafil
Some patients have trouble discussing problems relating to erectile dysfunction with their doctors, but it is important to step forward as erectile dysfunction can also be a symptom of other health problems such as clogged arteries or nerve damage. A doctor can offer a number of treatments for erectile dysfunction depending on the reason and underlying conditions.
While some treatments may involve a steady intake of medicines over a period of time, others can be as simple as taking a few pills for some days and getting more exercise and physical activity. The treatment generally lasts for about a month, but can also be of shorter or longer duration, depending on the severity of the disorder. If the erectile dysfunction is due to some other major ailment, then the problem generally subsides after complete recovery.
When a patient is suffering from erectile dysfunction, he generally has a very low self esteem and, hence, it becomes important that he get professional help and doesn’t try to deal with the situation all by himself. Becoming a part of a support group and taking psychological help from a psychiatrist often helps.
The most common medicines prescribed for erectile dysfunctions are sildanafil or viagra, vardenafil or levitra, and tadalafil or cialis. These medicines can cause side effects such as dizziness and headaches, and should be only taken under expert medical supervision. Some of the other side effects of these medicines may include an increased blood pressure and, thus, are not recommended for heart patients.
Remedies for Erectile Dysfunction
Here are several natural remedies that are used for erectile dysfunction.
L-Arginine
L-arginine is an amino acid that the body uses to make nitric oxide, a substance signals smooth muscle surrounding blood vessels to relax, which dilates the blood vessels and increases blood flow. Relaxation of smooth muscle in the penis allows for enhanced blood flow, leading to an erection.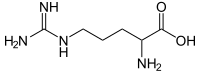
L-arginine is found naturally in foods such as meat, dairy, poultry and fish. It is also available as oral L-arginine supplements, which some product manufacturers market as a “natural Viagra”).
There have only been two studies to date, however, evaluating the effectiveness of L-arginine for erectile dysfunction.
One study involved 50 men who took L-arginine (5 grams a day) or a placebo. After six weeks, significantly more men taking L-arginine experienced an improvement in sexual function compared with men taking the placebo. Interestingly, it only benefited men who had initially low levels of nitric oxide.
Another study using a smaller dose of L-arginine and a shorter treatment duration found no benefit with L-arginine use. The study involved 32 men with erectile dysfunction who took oral L-arginine supplements (500 milligrams three times per day) or a placebo for 17 days. Oral L-arginine was no better than the placebo.
Side effects may include digestive complaints. High dosees of L-arginine may stimulate the body’s production of gastrin, a hormone that increases stomach acid. For this reason, L-arginine may be harmful for individuals with ulcers and people taking drugs that are hard on the stomach.
L-arginine may also alter potassium levels in the body, especially in people with liver disease. It should not be taken by people who are on medications that alter potassium levels, such as potassium sparing diuretics and ACE inhibitors
Propionyl-L-Carnitine
One study examined the use of two forms of carnitine, propionyl-L-carnitine and acetyl-L-carnitine in 96 men who with erectile dysfunction after prostate surgery. One group were given a placebo, another group took propionyl-L-carnitine (2 grams per day) plus acetyl-L-carnitine (2 grams per day) and sildenafil (Viagra) when needed, and the third group used Viagra alone.
Propionyl-L-carnitine and acetyl-L-carnitine were found to enhance the effectiveness of sildenafil, and result in improved erectile function, sexual intercourse satisfaction, orgasm, and general sexual well-being compared to Viagra alone.
Another study examined the effectiveness of propionyl-L-carnitine supplements plus sildenafil in men with erectile dysfunction and diabetes who were previously unresponsive to Viagra alone. Participants in the study received either propionyl-L-carnitine (two grams per day) plus Viagra (50 milligrams twice a week) or Viagra alone. After 24 weeks, propionyl-L-carnitine plus Viagra was significantly more effective than Viagra alone.
Gingko
The herb ginkgo is used for erectile dysfunction, particularly in people who experience sexual dysfunction as a side effect of antidepressant drugs. It appears to relax smooth muscle and enhance blood flow in the penis.

In one study of 60 men with erectile dysfunction, there was a 50 percent success rate after six months of ginkgo treatment. Two additional studies, however, found that ginkgo was no better than a placebo.
Zinc
Siginificant depletion of the mineral zinc, associated with long-term use of diuretics, diabetes, digestive disorders, and certain kidney and liver diseases, has been shown to lead to erectile dysfunction.
Ashwagandha

The herb ashwagandha (Withania somnifera) is sometimes called Indian Ginseng because it is thought to have similar effects on the body. It is thought to increase energy, stamina, and sexual function. No studies, however, have examined whether it is effective for erectile dysfunction in humans.
Side effects of ashwagandha may include drowsiness. It should not be combined with sedative drugs.
Yohimbe
The bark of the west African yohimbe tree is a source of yohimbine, a compound that has been found to stimulate blood flow to the penis, increase libido, and decrease the period between ejaculations.

Yohimbe is not recommended, however, because it is potentially dangerous, even in small doses. Side effects may include dizziness, anxiety, nausea, a severe drop in blood pressure, abdominal pain, fatigue, hallucinations, and paralysis.
Tongkat Ali
Tongkat Ali was dubbed the “Asian Viagra” in a May 1999 report in the New Sunday Times.
It has been used in Malaysia for many years by men to increase sexual desire, libido, sexual performance and to treat erectile dysfunction.

Tongkat ali appears to work by increasing levels of the hormone testosterone. Testosterone is primarily responsible for the growth and development of male reproductive organs, including the penis, testicles, scrotum, prostate, and seminal vesicles. Normal testosterone levels maintain energy level, mood, fertility, and sexual desire.
Because of its testosterone-enhancing properties, tongkat ali is also used by bodybuilders to increase muscle mass and strength
…………………………………………………………………………………………………..
Tribulus terrestris

Tribulus terrestris, also known as puncture vine, is a herb that has been used in the traditional medicine of China and India for centuries.
In the mid-1990s, tribulus terrestris became known in North America after Eastern European Olympic athletes said that taking tribulus helped their performance.
The active compounds in tribulus are called steroidal saponins. Two types, called furostanol glycosides and spirostanol glycosides, appear to be involved with the effects of tribulus. These saponins are found primarily in the leaf.
Tribulus is most often used for infertility, erectile dysfunction, and low libido. In the last decade, it has become popular to improve sports performance.
Tribulus has been marketed these conditions because research performed in Bulgaria and Russia indicates that tribulus increases levels of the hormones testosterone (by increasing luteinizing hormone), DHEA, and estrogen. The design of these research studies, however, has been questioned.
A more recent study found that four weeks of tribulus supplements (at 10 to 20 milligrams per kg of body weight daily) had no effect on male sex hormones testosterone, androstenedione, or luteinizing hormone compared to people who did not take tribulus.
Preliminary animal studies found that tribulus heightened sexual behavior and increased intracavernous pressure. This was attributed to increases in testosterone. There haven’t been any well-designed human studies to confirm these early findings.
…………………………………………………………………………………………………………………….
Maca
Maca (Lepidium meyenii) is a root plant consumed as a food and for medicinal purposes. Maca is also known as “Peruvian ginseng” (despite the fact that it is not a member of the ginseng family), because it is used as a folk remedy to increase stamina, energy, and sexual function. It is typically taken as a pill or liquid extract or as powdered maca root.
Long used to enhance energy and boost stamina, maca is often touted as an aphrodisiac and a natural means of improving sexual performance and fertility. Although few scientific studies have tested maca’s medicinal effects, some research suggests that maca may offer certain health benefits.
Proponents claim that maca may help with these health concerns:
- fatigue
- infertility
- symptoms of menopause
- sexual dysfunction in women
- sexual dysfunction in men (including erectile dysfunction)
Maca is also said to aid in the treatment of cancer.
Here’s a look at the available research on maca and its potential health benefits:
There is “limited evidence” for maca’s effectiveness in improving sexual function in men and women, according to a 2010 report published in BMC Complementary and Alternative Medicine. The report’s authors analyzed four clinical trials, two of which found that maca may have positive effects on sexual dysfunction or sexual desire in healthy menopausal women or healthy adult men. However, the other two trials found that maca failed to produce any positive effects on sexual function.
In a 2008 study from CNS Neuroscience & Therapeutics, researchers found that maca may help alleviate sexual dysfunction caused by use of selective-serotonin reuptake inhibitors (or SSRIs, a class of medications used in treatment of depression). The study involved 20 people with depression, all of whom were experiencing SSRI-induced sexual dysfunction. Results revealed that maca may also help improve libido.
………………………………………………………………………………………………………………
Muira Palma
Muira puama is a small Brazilian tree that grows across the Amazon river basin. It has a long history of use in Brazilian folk medicine as an aphrodisiac.
The root and stem of the tree are used medicinally.
Muira puama is used mainly as a herbal remedy for erectile dysfunction and sexual dysfunction in women.
……………………………………………………………………………………………………………..
Damiana
Other Names: Turnera diffusa, Turnera aphrodisiacaDamiana is a plant native to Mexico and the southern United States. The dried leaves are used medicinally.Damiana has been widely used as an aphrodisiac in Mexico for men and women.The use of damiana as an aphrodisiac is somewhat controversial because there is no scientific evidence that it works and yet it has been widely promoted as a sexual stimulant.One study suggests that damiana may have plant compounds with effects similar to those of progesterone. Over 150 herbs were tested for their ability to bind with estrogen and progesterone receptors in breast cancer cells and found that the damiana was among the six highest progesterone-binding herbs and spices.Damiana is also used for asthma, anxiety, depression, headache, and menstrual disorders, however, there is no scientific evidence that it works for these conditions.Damiana is found in various forms, including capsule, liquid extract, and tea form. A typical dosage is a 400 mg capsule taken once or twice a day.Damiana may cause mild indigestion.
Although damiana contains about 1/10 of the arbutin as the herb uva ursi, a maximum safe dose of damiana has not been established.
The safety of damiana in children, pregnant or nursing women, or people with liver or kidney disease has not been established.
…………………………………………………………………………………………………………..
Fo-Ti

Other Names: Polygonum multiflorum, He shou wu
Fo-ti is a plant native to China that is also found in Japan and Taiwan. The medicinal part of the plant is the root. In traditional Chinese medicine, it is often boiled in a liquid made with black beans — this is known as red fo-ti. White fo-ti is the unprocessed root.
Fo-ti is called He shou wu, which means “black-haired Mr. He” in Chinese. This name refers to a legend of an older villager named Mr. He who took fo-ti and restored his black hair, youthful appearance and vitality.
……………………………………………………………………………………………………………….
Horny Goat Weed
Horny goat weed is a leafy plant that is native to Asia and the Mediterranean region. It is also known as Epimedium and Yin Yan Huo.
Horny goat weed has a long history of use in traditional Chinese medicine.
According to folklore, horny goat weed’s reputed aphrodisiac qualities were discovered when a Chinese goat herder noticed increased sexual activity in his flock after they ingested the weed.
Animal studies indicate that horny goat weed may work by increasing nitric oxide levels, which relaxes smooth muscle and lets more blood flow to the penis or clitoris.
Horny goat weed also appears to act by inhibiting the PDE-5 enzyme, which is the same way that the popular drug Viagra works.
Some evidence suggests horny goat weed may modulate levels of the hormones cortisol, testosterone, and thyroid hormone, bringing low levels back to normal.
Share this:
]]>

E-Guggulsterone

Z-Guggulsterone
http://www.omicsonline.org/2161-0444/2161-0444-2-e105.php?%20aid=9899
Citation: Xiao M, Xiao D (2012) Gugulipid, an Extract of Ayurveda Medicine Plant Commiphora Mukul as a Potent Agent for Cancer Chemoprevention and Cancer Chemotherapy. Med chem 2:e105.
doi:10.4172/2161-0444.1000e105
Department of Urology, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania 15232, USA
| Gugulipid (GL), an extract of Commiphora mukul, has been safely used for thousands of years in the Indian Ayurveda medicine practice for the treatment of different ailments and has been used recently in many clinical trials that focused on its cholesterol-lowering effect. GL has recently been paid great attention for its cancer chemopreventive and chemotherapeutic potential. Z- and E-Guggulsterone have been identified as the major active components of GL. Studies have shown that GL as well as Guggulsterones can inhibit cancer growth in vitro and in vivo and lead to prevention of cancer initiation, promotion and progression. Although the action mechanisms of GL are not completely understood, GL has been revealed as a multitargeted cancer chemopreventive and chemotherapeutic agent. The increased understanding of the anti-cancer activity of GL and its molecular targets would allow us to improve its efficacies in different types of human cancers by single and/or combination strategies. | |||||
Gugulipid (GL, guggul, guggal, or gugul lipid) is the ethyl acetate extract of gum guggul resin (raw material) that is harvested directly from the Commiphora mukul tree (family name: Burseraceae; synonyms: Hook, Bandari, Balsamodendron mukul, and Commiphora Wightii). GL is a highly valued botanical medicine. As aforementioned, GL has been safely used for thousands of years in the Indian Ayurvedic medicine for the treatment of different ailments, including lipid disorders, rheumatoid arthritis, ulcers, osteoarthritis, bone fractures, epilepsy and obesity . In 1986, GL was granted approval in India for marketing as a lipid lowering drug (Indian Pharmacopeia 2007: pgs. 2038-2040). Several products of standardized formulations of Commiphora mukul are already in human use as cholesterollowering agents . The Z- and E-forms of guggulsterone (Gug, 4,17(20)-pregnadie-3, 16-dione) have been identified as major active components of GL . GL and its active component z-Gug have been used in many clinical trials that focused on its cholesterol-lowering effect . Numerous studies continue to support that many edible phytochemicals have cancer chemopreventive and chemotherapeutic potential . These finding motivated the investigation of the cancer chemopreventive and chemotherapeutic potential of GL and its active components.Guggulsterone is a plant steroid found in the resin of the guggul plant, Commiphora mukul. Guggulsterone can exist as either of two stereoisomers, E-guggulsterone and Z-guggulsterone. In humans, it acts as an antagonist of the farnesoid X receptor, which was once believed to result in decreased cholesterol synthesis in the liver. Several studies have been published that indicate no overall reduction in total cholesterol occurs using various dosages of guggulsterone, and levels of low-density lipoprotein (“bad cholesterol”) increased in many people.[1][2] Nevertheless, guggulsterone is an ingredient in many nutritional supplements.
Scheme 1. Stereoselective synthesis of E-guggulsterone (1) from 4-androsten-3,17-dione (5). (i) CH(OEt)3, p-TSA, THF/EtOH, quantitative; (ii) EtPPh3Br, t-BuOK, THF, reflux; (iii) HCl, THF, 95% from 6; (iv) SeO2, t-BuO2H, 90%; (v) (COCl)2, DMSO, Et3N, CH2Cl2, 85% |
|||||

Share this:
]]>